 加载中…
加载中…NCBI下载SRA数据和之后的数据处理
| 分类: 生信软件使用 |
首先是如何下载:
需要安装SRA toolkit:
https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc&f=std
以下主要是在linux系统中的操作。
再根据所查到的每一个run的序号,如SRR**(或DRR**)下载数据,可以利用命令prefetch
SRR**,需要注意的是,必须是
SRR开头!
将下载的数据转换成为合适的格式:
(首先cd到数据的存放地,如cd ~/ncbi/public/sra)
fastq格式--~/sratoolkit2.8.0-ubuntu64/bin/fastq-dump
sff格式--~/sratoolkit2.8.0-ubuntu64/bin/sff-dump
其次是如何将SRA原始测序数据转为fastq格式
一,下载该软件
wget
tar
解压直接使用即可,里面有一大堆的软件,针对不同的测序仪,不同的数据
http://www.bio-info-trainee.com/wp-content/uploads/2015/03/SRA%E5%B7%A5%E5%85%B7sratoolkit%E6%8A%8A%E5%8E%9F%E5%A7%8B%E6%B5%8B%E5%BA%8F%E6%95%B0%E6%8D%AE%E8%BD%AC%E4%B8%BAfastq%E6%A0%BC%E5%BC%8F448.png
{kind=link}
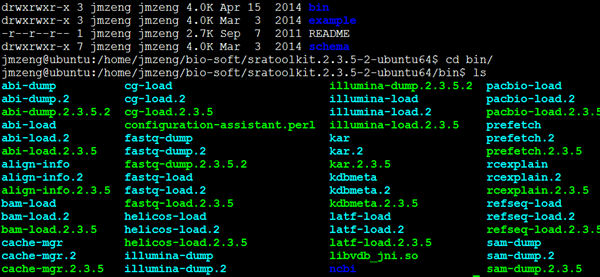
我一般只用/home/jmzeng/down_software/sratoolkit.2.3.5-2-ubuntu64/bin/fastq-dump
/home/jmzeng/down_software/sratoolkit.2.3.5-2-ubuntu64/bin/fastq-dump
二:下载数据
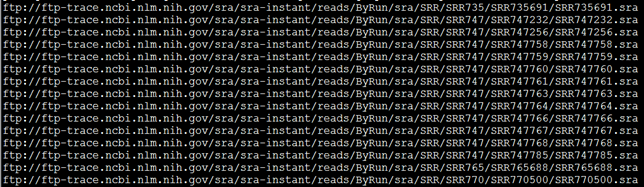
首先去NCBI里面搜索并找到你想要的数据的SRA地址,然后写脚本批量下载。
http://www.bio-info-trainee.com/wp-content/uploads/2015/03/SRA%E5%B7%A5%E5%85%B7sratoolkit%E6%8A%8A%E5%8E%9F%E5%A7%8B%E6%B5%8B%E5%BA%8F%E6%95%B0%E6%8D%AE%E8%BD%AC%E4%B8%BAfastq%E6%A0%BC%E5%BC%8F826.png
{kind=link}
如果文献里面的SRA号,那么可以直接打开NCBI里面的搜索界面下载
如果文献里面是SRP号,那么该SRP会涉及到好几个SRA数据,得一个个开网站下载
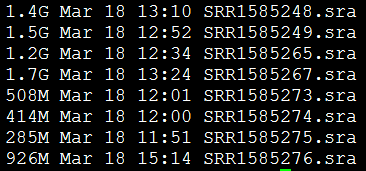
三:用命令解压数据
下载之后的数据是
http://www.bio-info-trainee.com/wp-content/uploads/2015/03/SRA%E5%B7%A5%E5%85%B7sratoolkit%E6%8A%8A%E5%8E%9F%E5%A7%8B%E6%B5%8B%E5%BA%8F%E6%95%B0%E6%8D%AE%E8%BD%AC%E4%B8%BAfastq%E6%A0%BC%E5%BC%8F1008.png
{kind=link}
非常简单的命令,就可以把当前文件夹下的所有sra都解压开来!
[shell]
for i in *sra
do
echo $i
/home/jmzeng/bio-soft/sratoolkit.2.3.5-2-ubuntu64/bin/fastq-dump
--split-3 $i
done
[/shell]
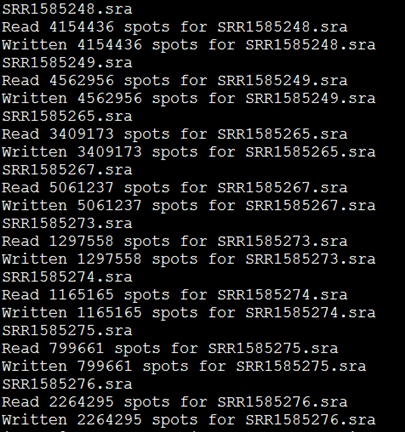
解压的同时它也会显示每个SRA文件的数据量
http://www.bio-info-trainee.com/wp-content/uploads/2015/03/SRA%E5%B7%A5%E5%85%B7sratoolkit%E6%8A%8A%E5%8E%9F%E5%A7%8B%E6%B5%8B%E5%BA%8F%E6%95%B0%E6%8D%AE%E8%BD%AC%E4%B8%BAfastq%E6%A0%BC%E5%BC%8F1064.png
{kind=link}
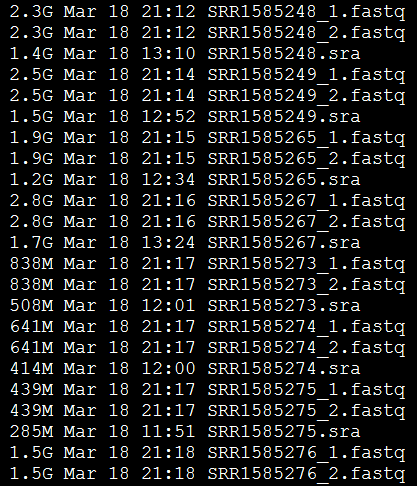
四:结果文件解读
http://www.bio-info-trainee.com/wp-content/uploads/2015/03/SRA%E5%B7%A5%E5%85%B7sratoolkit%E6%8A%8A%E5%8E%9F%E5%A7%8B%E6%B5%8B%E5%BA%8F%E6%95%B0%E6%8D%AE%E8%BD%AC%E4%B8%BAfastq%E6%A0%BC%E5%BC%8F1235.png
{kind=link}
可以看到,每个SRA文件都产生了两个reads,分别是左右两端测序,说明这个SRA文件是双端测序策略。
随便打开一个fastq文件可以看到,它的读长是300bp
http://www.bio-info-trainee.com/wp-content/uploads/2015/03/SRA%E5%B7%A5%E5%85%B7sratoolkit%E6%8A%8A%E5%8E%9F%E5%A7%8B%E6%B5%8B%E5%BA%8F%E6%95%B0%E6%8D%AE%E8%BD%AC%E4%B8%BAfastq%E6%A0%BC%E5%BC%8F1479.png
{kind=link}
![]() 喜欢
喜欢
0
![]() 赠金笔
赠金笔