 加载中…
加载中…差分电荷密度图、电荷局域密度图(ELF)的画法及分析2
| 分类: 科研日志 |
转载自
处理步骤:【需要商议的步骤】---另一种定义电荷密度差
http://muchong.com/html/201512/9799282.html
用VESTA 做VASP差分电荷(C-A-B)图方法:
1、导入 C 的CHGCAR(拖拽到vesta)面板框中
2、Edit——>Edit data——>Volumetric Data————>Import (选择A
的CHGCAR)——>Subtract
from current data——>OK
3、Edit——>Edit data——>Volumetric Data————>Import (选择B
的CHGCAR)——>Subtract
from current data——>OK
注意:A,B,C三个文件的格点要一一对应。
文献分析:
(1)Cr合金化Mg_2Ni氢化物能量与电子结构的第一性原理研究
的差分电荷密度图(是指原子---组成体系(团簇)之后电荷的重新分布, 可以很清楚地看出体系中原子的成
键情况)如图4 所示。
http://image.sciencenet.cn/album/201608/18/110838ddxwii5dsq5a8sld.png
{kind=link}
(2) 放热型金属合金化对钒基贮氢材料性能影响的理论研究
在氢化物 VH2
(3)金属氢化物力学性能的第一性原理研究
http://image.sciencenet.cn/album/201608/18/114300bqzkf9zfkq4iqk9k.png
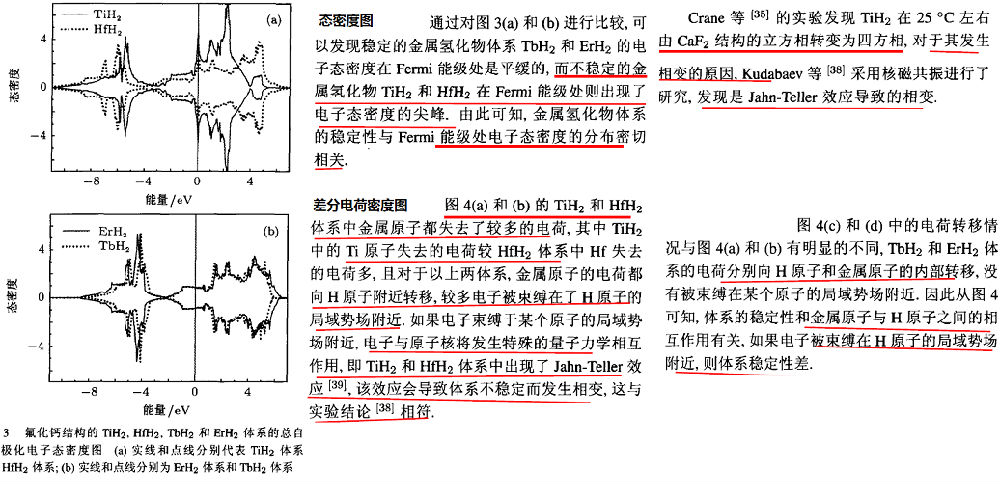{kind=link}
http://image.sciencenet.cn/album/201608/18/115701yodetz06osydjetc.png
{kind=link}
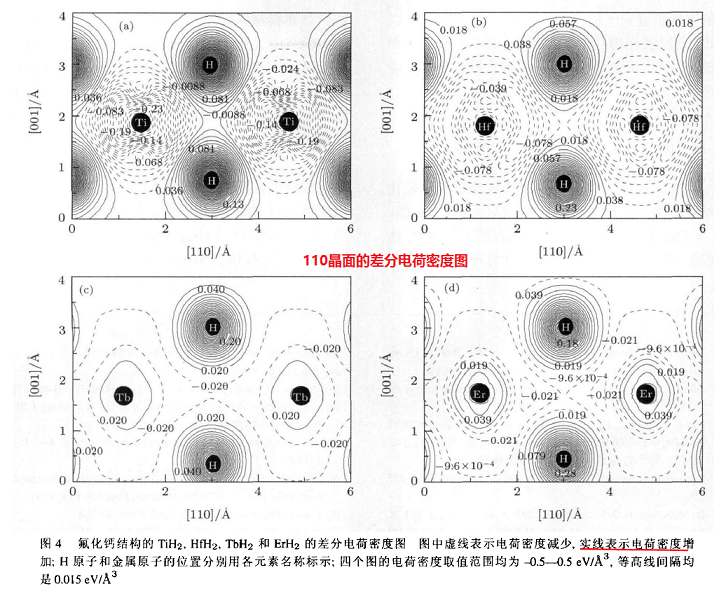
(4) Hydride-Based Electride Material, LnH2 (Ln = La, Ce, or Y)
Figure 3a shows the
electron
{kind=link}
(5) 差分电荷密度图--透明钠:Transparent dense sodium
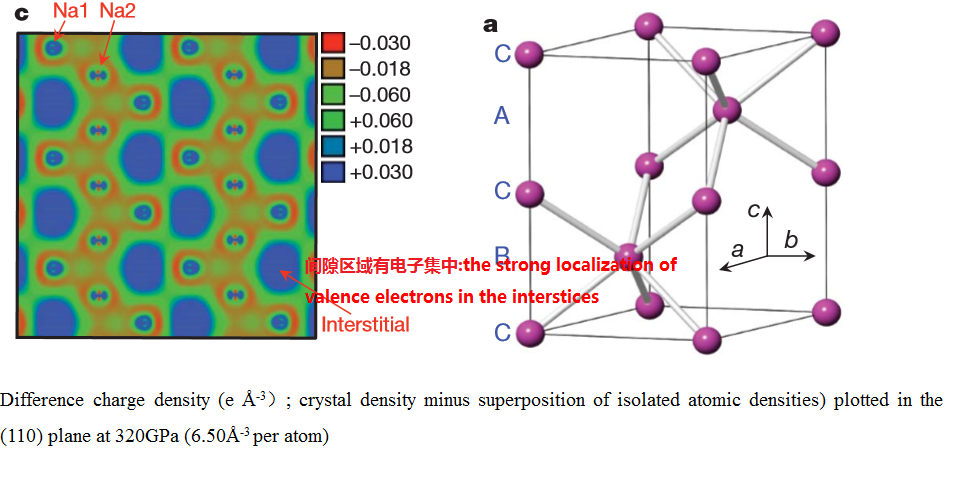{kind=link}
Difference charge density
(e
Thus,
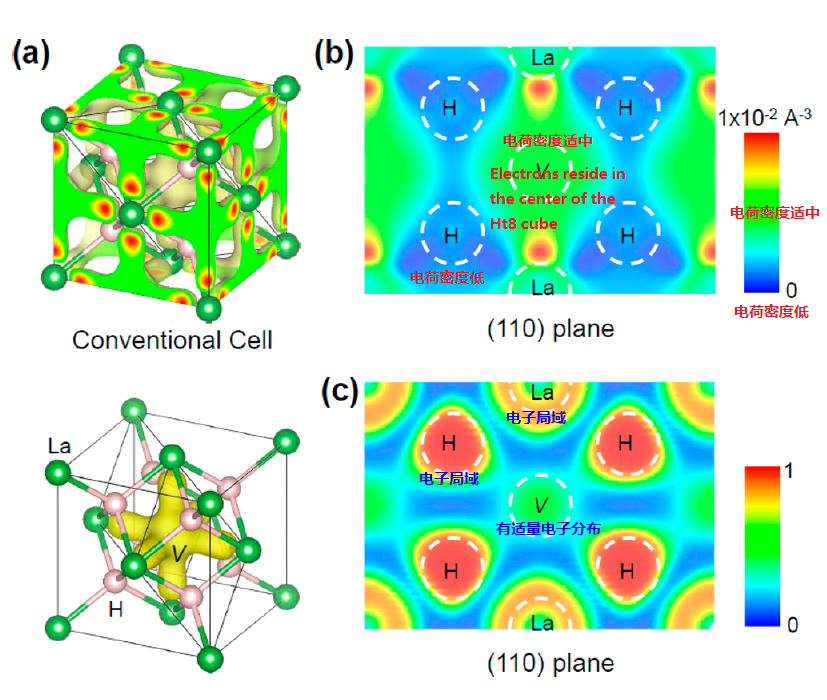
(5) Pressure-stabilized superconductive yttrium hydrides
http://image.sciencenet.cn/album/201608/20/124155ivrr98arrv0vapks.png
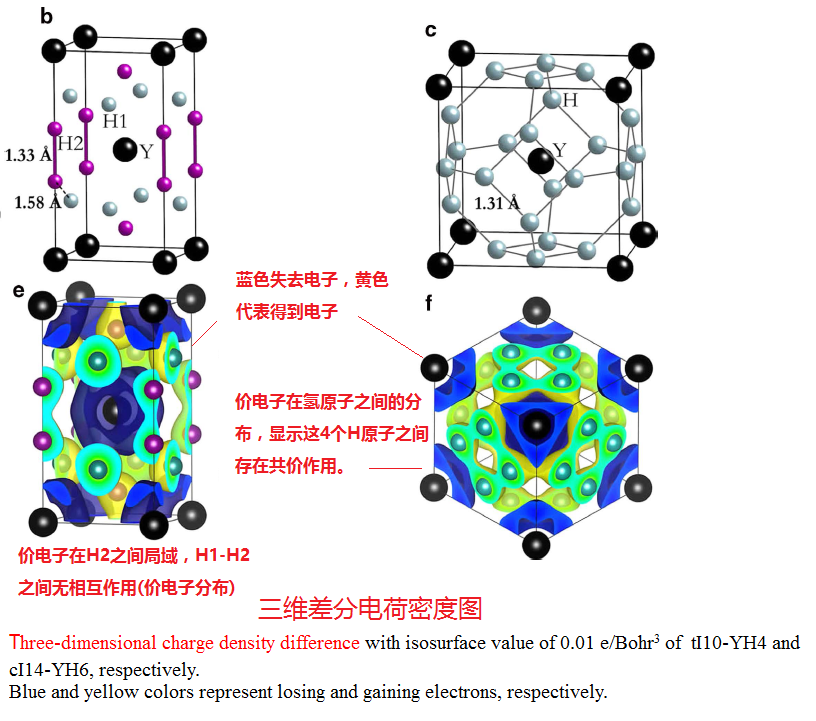{kind=link}
Valence electrons localization was found
Despite the longer
distance,
http://image.sciencenet.cn/album/201608/20/125606klwz945swetbeb9w.png
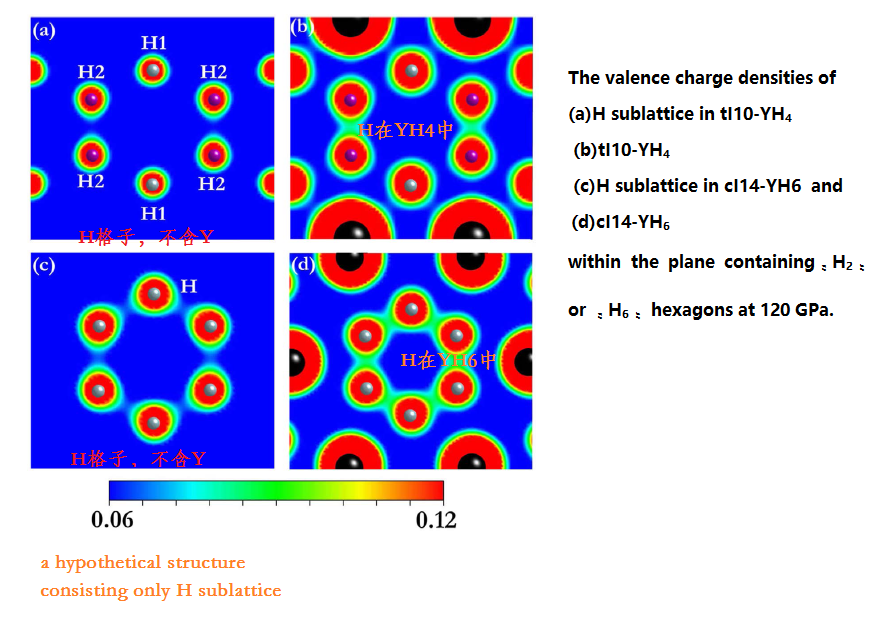{kind=link}
http://image.sciencenet.cn/album/201608/20/13354186tkkzcmkmmxxr11.png
{kind=link}
网络问答:
(1) AA::
如何在vaspview中画电荷密度等高线,图是我画出的电荷密度,想在上面添加等高线,求大能帮助
vesta:
Utilities->2D Data
Display->Coutour
(2) AA::
怎么调节切片方向呢? 我切出来是这样的,调节ranges of
fractional coordinates中参数都不管用,是不是调节其他参数啊
QQ:::
Utilities->2D Data
Display->Coutours->Slice->hkl
然后还有distance from origin可以调
[距离原点的距离?]
QQ:::
用lev00来分析也很好。。。比较方便。
对的,你安装好lev00后,里面会有很多选项,包括可以任意指定切面。。
然后再用origin画。
(3)
比较了一下,感觉还是vesta出来的图美观和操作方便些。vaspview里面的功能太少了。vesta切slice也很方便,不管z轴还是x轴还是任意方面的面都可以切。vesta中你的线消失是因为你切的面没落在原子上当然等高线就出不来了。
QQ:::可切任意面,注意切面到原点的距离,就那么几项,仔细看看,你一定能搞定
终于搞定了,原因出在CHG与CHGCAR格式的差异上,原来做差时用的是CHGCAR,差值也按CHGCAR的格式保存的,但是VESTA认的是CHG的格式,用CHGCAR的格式就出现了上述情况,现在按CHG格式保存电荷密度差,结果就正常了。
(5)
第一个问题就可以在MS中把晶胞调整参数后rebuilt一下就可以了;第二个问题建模用MS的话就更方便了,建晶胞--切表面--做真空层--建超胞
QQ:::
(6)
能不能告诉我具体第几章第几节谈到了为什么不能导出2D电荷密度数据的问题?
我看了Manual上面13章仅仅一笔带过说这个选项是导出数据。
QQQ:::"15.7
Exporting 2D data
To export 2D data of the specied slice as a text le, choose File"
menu ) Export 2D Data. . . "
in the 2D Data Display window. No 2D data les can be output on
selection of style (hkl) plane
in the bounding box" in the Slice Properties dialog
box."
(7)
(8)
【如何把Slice图放到结构当中,像下面这样?】
http://blog.sciencenet.cn/static/js/grey.gif
{kind=link}
QQQ:::可以呀 在2D视图里面,可以设置等高线和色彩的
QQQQ3::::
http://blog.sciencenet.cn/static/js/grey.gif
看到那个saturation levels
吗?最大的是2.98023(100%),最小的是0.5871(0%),而右边的标尺是根据那个把最大和最小分成十份,你自己算一下标上去就行了
(9)
【如何把Slice图放到结构当中,像下面这样?】
http://blog.sciencenet.cn/static/js/grey.gif
QQQQ::
这个图可能不是vesta做的
对于这个图可以这么做:
1,先从2D里面吧标尺弄出来放在图片一中,,
2,然后在boundary里面设置晶体的晶面,返回带properties里面选择isosurfaces,在这里修改isosurface
level为0(里面的颜色不会随你的这个值而改变,设置为零(你的电荷的最低值,也就是你标尺最下端得值)一般能把所有的数据显示出来),这样可以得到图二,
3,返回主界面,把Volumetric data 里面的
show section前面的钩去掉,再到properties里面选择isosurfaces,在这里修改isosurface
level为设置为你想要的值,但此时这个颜色会和那个标尺不一样(默认的是黄色)。你可以现在标尺中找出你想要值的颜色,使用其他的程序读取RGB值,然后再在color中填上就可以。如果你都不了那个RGB值的话,就靠你的眼睛去判断,点后面那个按钮。可以选择你认为是一样的值。得到图三
4,在画图版中,使用那个白色透明模式(工具栏中最下面有两个大的黄色的下面的一个),把这几张图合并起来就行。
在整个过程中你不要去调图像看看图像怎么样,或者你看好之后用orientation,回到以前的视图角度,这样可以保证两张图是可以完全重合的
(10)空位、间隙原子 密度图的处理
(a)vasp算出CHGCAR后可以导入VESTA,给出整个超胞的电荷密度图,但我只关心其中某几个原子附近的电荷密度分布,求教高手,能否只显示我关心的这几个原子上的电荷密度,如果能够,如何实现?
(b)超胞计算中引入了空位,能否在图示中用虚线球给出该空位的理想位置,并且用虚线表示出其本来的成键情况,如果能够,如何操作?
非常感谢!
QQQ:::vesta
似乎是没有办法只显示几个原子的电荷密度的,只关心几个原子,是不是可以考虑差分电荷分析?就是把你要关注的几个原子作为一个体系(去掉其他原子,仍用原来晶格)保持坐标不动,单独算一下,得到CHGCAR1,
然后把其他部分的原子(去掉关心的原子)保持坐标不变再单独算一下,得到CHGCAR2,
然后用总体系的CHGCAR-CHGCAR1-CHCAR2 得到CHGCAR——difference。 然后再用vesta
做CHGCAR-difference。但和你的要求可能有点区别。
引入空位的话,vesta
似乎不能做到你的要求,但是你可以在vesta里面作结构图,然后导出图片,在ppt或者Photoshop
中修改图片。可以得到满足你要求的图片
QQQ::::
只显示几个原子而不显示其他原子的电荷密度很奇怪的,没有太多意义。我建议你采用截面的方法来显示某几个原子附近的电荷密度分布
(11)再用VESTA画部分电荷密度(partial
charge density)的时候,isosurface level
一般取值为多少。有什么原则吗,比如最大值的多少分之一。我刚刚学不清楚。请高手指点下,提前谢
(12)A:::选择适当的平面使要观察的原子的ELF落在这个平面上
在用VESTA做ELFCAR的2D图的时候要选择适当的平面使要观察的原子的ELF落在这个平面上,我的问题是用VESTA作2D图时显示不出原子,那么怎么自己加原子呢?比如下面两个图:图1是3D图,其中紫色的是Zn,灰色是O,黄色是cu,图2选取的001面,那么在图2的那些区域,怎样确定那个是In产生的哪个是粗cu产生的呢?
又或者:怎样让001面上只显示出cu及其周围的Zn和O?
Q:::记得slice里面不仅可以选哪个面,还可以定义面的位置,比如在最底下(z=0),或者中间(z=0.5)……
应该是可以定义的。
你图片上看到“slice”这个键了吗?点击就出现那个对话框里,h、k、l 三个指数定义面,下面有个“Range of
projection”,计算定义原点到面的距离,一个的以angstrom为单位,另一个是分数坐标吧。
不知道怎么回事,上传不了图,不好意思啊。
(13)电荷密度图单位问题:CHGCAR的单位应该是e,
我想问的是有没有简便的设置不用每次都这样改?太麻烦了!
QQ::
问一下楼主,你从哪里知道CHGCAR的单位就是e/Bohr^3呢?
我最近看了一些相关资料知道,CHGCAR的单位应该是e,
VASP说明书上有这样一句话:
“CHGCAR
file
This file contains the
lattice vectors, atomic coordinates, the total charge density
multiplied by the volume
“ Please do not forget to
divide by the volume before visualizing the
file!”
(14)
差分电荷密度的另一种算法:Δρ=ρ(AB)-ρ(A)-ρ(B)
Q:::
会不会是因为INCAR里面某些参数是使用默认值,而这些值是根据POTCAR读来的,结果因为POTCAR不同而导致INCAR也发生了变化?比如ENCUT是可以不写的,如果3个INCAR里面都没有这一行,而是采用PREC=Accurate的设置的话,那么AB那一次计算的ENCUT会跟ENMAX比较高的那个(比如B)的一致,而ENMAX比较低的那个(比如A)算出来的就是另一回事了。不妨把那些可以手动设置的参数自行设置一下,比如ENCUT、NGX、NGY、
NGZ和NGXF、
NGYF、NGZF之类的…当然我理解也不是很深,不确定是不是这种情况。
VESTA软件的用法讨论:摘自大家identation:
http://emuch.net/bbs/viewthread.php?tid=6422652&fpage=1
请大家交流一下VESTA软件的用法
这个取值是你自己取得,就看画出的图能不能描述出问题的所在,比如你中心原子电荷是0.5,外部空间是0.2,那你就不能将0-1取为一个颜色,因为这样你无法分辨外部中心原子和外部空间。VESTA作图的单位我还真不清楚,我做密度图的时候都是用lev00然后导出数据用origin画的,不过好像VESTA的单位和VASP的单位不太一样,这个你自己查一下
a)首先,VESTA的作用可以是--画晶体结构,当你知道了晶体的空间群,原子的Wyckoff坐标之后,就可以用VESTA画结构了:
(1)File->New Structure->Unit cell->Space group处输入空间群的编号,Setting处可以对该空间群编号下的不同表述方式进行调整,例如No. 62号空间群属于正交晶系,输入62在Space group处之后,如果setting是1,则显示为Pnma,如果setting为3,则显示为Pbnm,实际上这两个空间群是等价的。
(2)接下来,structure parameters处,点击New,然后在Symbol处选择你想输入的元素,Label处输入你想显示的标签,同时把x,y和z后面的空格处填上该元素的坐标,则该原子输入完毕,再点击New,输入其他原子坐标,直至完成。
(3)完成之后,你将看到你所画的晶体结构图。
(4)对于该图,你可以有多种显示方式,例如键线式,八面体或四面体等等。这需要如下步骤来做到--点击Edit->Bonds->点击New,然后将A1和A2处填上成键的原子,例如考虑体系BiFeO3,如果想显示Bi-O八面体,则可把A1处的原子写成Bi,A2处写成O,然后注意调整Min. length和Max. length处的数值,这两个值的意思是,如果A2原子到A1原子的距离在Min. length和Max. length数值之间,则认为这两个原子成键,两原子直接连一线。然后点击OK即可。
(5)如果要八面体或者四面体显示该晶体,在完成前4步之后,点击Style按钮,选上Polyhedral,则可以以多面体显示。
(6)Objects->Properties处可以改变显示的风格--例如可以改变多面体的颜色,可以使用超晶报显示等等。
此外,也可以将所画晶体用图片格式导出,可以导出矢量图也可以导出普通的图片,请点击File->Export Raster Image或File->Export Vector Image
(b)VESTA可以显示电荷密度,
(2)Utilities->2D data
display
(3)你同样可以试着在Edit->Lattice planes处做类似操作,获得在原晶体结构上显示的某一晶面的2D电荷密度投影。
(c)改变基矢:
最最简单的玩法是,当你把Rotation matrix
P第三行第三列的那个数值由1改成2的时候,你将获得z方向的超晶报;同理如果把第二行第二列的数值由1改为2,你将或者y方向的超晶胞。
VESTA是一个功能很强大的晶体建模显示工具,我个人认为其基本上可以囊括MS的可视化模块,而且显示起来的图更好看。也希望大家多多交流,说说你所知道的VESTA的用法或作用。有金币悬赏!
(d) 講一些關於
isosurfaces 分析:
1)
2) 在
isosurfaces 分析過程有許多參數可以調整,東調西調~調完一大堆參數後
4) 把 CHGCAR 丟入 VESTA 之後
5)
VESTA画局域电荷密度图 ELF
http://muchong.com/html/201508/9267921.html
VESTA可以切片
Utilities---2D DATE
就在VESTAT中调整啊
1、体材料上,调整Isosurface level
2、切面:只能调整显示
电子局域函数源表征:电子的局域化分布特征:用于确定成键类型和找出孤对电子分布的理由。
http://blog.sina.com.cn/s/blog_6813cb430100tvka.html
差分不同的定义:
http://muchong.com/html/201607/10515384.html
(1)
deformation charge density
这个的定义是用总的电荷,减去体系中原子电荷,就是他问的你这个电荷是非自洽电荷,is the non-self-consistent
charge density
(2)
(2) 差分电荷密度从Gaussian的解释
http://blog.sina.com.cn/s/blog_76cd5d7c0102vnzl.html
对于G03来说,它的cubegen是能作density和Orbitals等的空间格点的。
但是它的density(ρ)只是全分子的,分子的所有MO的电子密度加和,还自然包含分子中的1S、2S轨道,用图形展示出来是一个圆滚滚的样子,很难说明什么,也不利作密度差。
而G03的cubegen不能作某个MO的ρ,将某个MO的cube输入到cubman也不能作平方运算作成ρ,因为cubman缺少平方运算。
gsgrid程序附有详细的使用说明,一看就会。
而进入高超的sigmaplot中作等值线,更是功能强大、得心应手!
具体计算如下:
单个原子的AO的获得,要单算。
如除了算HF外,H和F还要另外单算,而且它们要与HF有同样指定的空间格点规格,且与HF中的H和F有相同的坐标位置,相同的基组。
如果是算整体的ρ来相差,这样得到的三个cube,进入gsgrid作三个xy截面G1、G2、G3,再用HF的G1减去H和F的G2、G3,即得电子密度差。进入sigmaplot作图、调整即得等值线图。
如果想作精细一些,需要在HF中指定一个MO来作,如上面的σ键的MO,它是第3个MO。F也要选定那个单电子的AO,H就是一个了。用指定的MO作三个cube,进入gsgrid作三个xy截面G1、G2、G3,再对这三个xy截面分别作平方使成为电子密度,HF的这个轨道有2个电子,还要乘以2。再用HF的减去H和F的,即得电子密度差。进入sigmaplot作图、调整即得等值线图。
实践表明,因为这时由原子到分子,真正发生电子净变化的主要是σ成键轨道。内层F1s等基本没有变化,相差之后抵消了,而F的孤对电子轨道,虽然因为通过σ键诱导过来部分电子,对有效核电荷的屏蔽加强,使核对孤对电子pi吸引能力减小、能级升高,表现为轨道膨胀、弥散,但由于我们的xy截面是对σ键的中心剖面,对这个截面,孤对电子pz与之是正交的、xy截面是其节点,没有截取到值,另一孤对电子py也因弥散影响甚微。所得到的电子净变化图形,与上面根据σ成键轨道MO作出它的没有原则性的差别。
![]() 喜欢
喜欢
0
![]() 赠金笔
赠金笔