 加载中…
加载中…【佳学基因检测】ADCA-DN综合征基因解码、基因检测
标签:
基因检测基因解码佳学基因adca-dn综合征 |
遗传病、罕见病基因解码基因检测导读:
ADCA-DN综合征基因检测是对英文疾病名称为ADCA-DN Syndrome的疾病所进行的基于序列变化的分子诊断手断。该病的其他英文表达方式还有:Autosomal Dominant Cerebellar Ataxia, Deafness and Narcolepsy、ADCADN、Autosomal Dominant Cerebellar Ataxia-Deafness-Narcolepsy Syndrome、Autosomal Dominant Cerebellar Ataxia, Deafness, and Narcolepsy、ADCA-DN Syndrome、 Autosomal Dominant Cerebellar Ataxia-Hearing Loss-Narcolepsy Syndrome、Ataxia, Cerebellar, Deafness, and Narcolepsy, Autosomal Dominant、ADCA-DN。因此,ADCA-DN综合征基因检测又以常染色体显性小脑性共济失调、耳聋和发作性睡病基因筛查、 ADCADN基因测主式、 常染色体显性小脑性共济失调-耳聋-发作性睡病综合征遗传性分析、 常染色体显性小脑性共济失调、耳聋和发作性睡病致病基因鉴定、 ADCA-DN综合征遗传性测试、 常染色体显性遗传性小脑性共济失调-听力损失-发作性睡病综合征致病基因鉴定基因解码、 共济失调、小脑、耳聋和发作性睡病,常染色体显性遗传全基因测序分析、 ADCA-DN全外显子测序检查的形式出现。
常染色体显性遗传性小脑性共济失调、耳聋和发作性睡病 (ADCA-DN) 是一种影响神经系统的罕见遗传病。 它的特征是三联征,包括小脑性共济失调、感音神经性听力损失和发作性睡病伴猝倒。
小脑性共济失调是一种影响运动协调和平衡的神经系统疾病。 患有 ADCA-DN 的人会出现步态不稳、精细运动任务困难和言语不清。 感音神经性听力损失通常始于儿童早期,并随着时间的推移而发展,导致某些人完全失聪。 发作性睡病伴猝倒症是一种睡眠障碍,会在情绪激动时引起肌肉无力或瘫痪的突然发作。
ADCA-DN 是由 DNMT1 基因突变引起的,该基因编码一种参与 DNA 甲基化的酶,这是一种调节基因表达的过程。 DNMT1 的突变导致酶的结构和功能发生变化,从而破坏 DNA 甲基化和基因表达,导致 ADCA-DN 的症状。
ADCA-DN 以常染色体显性遗传模式遗传,这意味着只需要一个突变基因拷贝即可发展该病症。 父母患有 ADCA-DN 的人有 50% 的机会遗传该病症。
目前,ADCA-DN 无法治愈,治疗的重点是控制症状。 物理治疗可以帮助解决步态和平衡问题,而助听器可以改善听力。 兴奋剂和抗抑郁药等药物可以帮助控制发作性睡病伴猝倒症。 还建议对受 ADCA-DN 影响的个人和家庭进行遗传咨询。
什么样的人应当做ADCA-DN综合征基因检测基因解码?
常染色体显性遗传性小脑性共济失调、耳聋和发作性睡病 (ADCA-DN, MIM 604121) 是一种多态性病症,于 1995 年首次在瑞典谱系中描述,其中鉴定并研究了五个受影响的个体 。该疾病的特征是晚发(30-40 岁)发作性睡病-猝倒、感觉神经性耳聋、小脑性共济失调、痴呆,以及更为多变的精神病、视神经萎缩和其他症状。脑脊液 (CSF) hypocretin-1 的研究表明其水平较低或检测不到,这表明这些受试者发作性睡病的原因是产生 hypocretin 的细胞缺失。
佳学基因检测收集另外三个病例:(i) 来自美国的大型多代常染色体显性遗传家系,有 13 名受影响的个体,其中 6 名活着;(ii) 该病在一名 50 岁的意大利患者中偶发,其父母未受影响,表明存在新发突变;(iii) 具有 4 个已知受影响且具有相似病程的多重意大利血统。意大利的零星病例值得注意,因为发作性睡病 - 猝倒症是第一个症状(42 岁),其次是听力丧失、记忆问题和抑郁症(43 岁)、下肢淋巴水肿(45 岁)、小脑性共济失调(46 岁)、外周感觉神经病(47 岁)和视神经萎缩(55 岁)。CSF hypocretin-1 水平较低(表 1个),CSF tau 蛋白高(614 pg/ml,正常值 141 ± 127 pg/ml),而 14-3-3 水平正常。多次睡眠潜伏期测试 (MSLT) 也呈阳性。与原始瑞典谱系 ( 1 ) 的某些成员一样,HLA-DQB1*06:02 在两个新的意大利谱系中均为阴性,除了一个受影响的美国亲属外。在所有这些病例中,发作性睡病 - 猝倒是一种早期症状,尽管发病年龄和症状发生率存在差异,正如瑞典家庭所报告的那样 (1) (见表 1个有关先证者的代表性数据,包括可用时的 CSF hypocretin-1 水平)。为了确定 ADCA-DN 的病因,基因解码基因检测对三个家系的五个受影响成员进行了外显子组测序。在确定DNMT1导致该疾病后,还在所有四个亲属的所有可用样本中对DNMT1基因进行了测序。
表1:佳学基因ADCA-DN 综合征病例介绍
| 表型 | 病例1 | 病例2 | 病例3 | 病例4 |
|---|---|---|---|---|
| 突变 | Ala570Val | Ala570Val | Gly605Ala | Val606Phe |
| 性别 | 男 | 男 | 男 | 男 |
| 年龄 | 57 | 58 | 47 | 56岁(已故) |
| 白天过度嗜睡 | 42 | 35 | 43 | 18 |
| 猝倒发作 | 42 | 44 | 43 | 32 |
| 睡眠麻痹 | 无 | 无 | 无 | 有 |
| 入睡幻觉 | 无 | 无 | 有 | 有 |
| REM睡眠行为障碍 | 有 | 有 | 有 | 有 |
| MSLT(SL、SOREMP) | 5, 5 (47岁) | 2, 4 (45岁) | 10 岁,4 岁(47 岁) | 11 岁,3 岁(29 岁) |
| 小脑性共济失调 | 46 | 48岁 | 47 | 37 |
| 听力障碍 | 43 | 48岁 | 43个 | 32 |
| 记忆力减退和抑郁 | 43 | 52 | 没有 | 是的 |
| 四肢淋巴水肿 | 45 | 没有 | 47 | 没有 |
| 感觉神经病 | 47 | 55 | 47 | 49 |
| 视神经萎缩 | 55 | 57 | 47 | 38 |
| II型糖尿病 | 没有 | 38 | 没有 | 47 |
| 失智 | 没有 | 54 | 没有 | 52 |
| 精神病 | 55 | 52 | 没有 | 47 |
| Hypocretin-1 (pg/ml) | 123 | NA | 93 | 62 |
| HLA-DQB1*06:02 | 阴性 | 阴性 | 阴性 | 阴性 |
ADCA-DN综合征基因检测结果
外显子组捕获和测序是在散发的意大利病例加上他未受影响的父母,以及来自瑞典和美国的两个由多个成员组成的家系进行的。每个家系中有两个患者。对于每个样本,在
HiSeq 2000 系统上对流通池的 0.33 条泳道进行测序,平均每个样本生成 8.77 Gb 的序列。96.5%
的基因序列可以定位参考基因组 (hg19),平均 80.2% 基因测序序列定位到外显子组基因编码序列。平均读取深度为 108,其中
88% 至 91.5% 的目标区域至少覆盖了 20 次。使用 Burrows-Wheeler 比对 (BWA) (v 0.5.9)
进行读取比对并使用 SAMtools 通过基因解码分析流程识别单核苷酸错义、无义、剪接位点、终止和移码变异以及小的插入和缺失
(indel) (v
0.1.7),从而获得受检者的基因突变列表。基因解码对这些变异进行过滤、筛选,以排除
使用这种策略,基因解码发现瑞典家庭的两个同为患者的兄弟姐妹共有 77 个个人独特性变异,而美国家系的两个表亲共有 30个体独特性变异
个。在意大利先证者中,致病基因鉴定基因解码确定了两个被确定为从头发生的错义变异。在所有五个患者中,共同发生的突变只有一个基因DNMT1
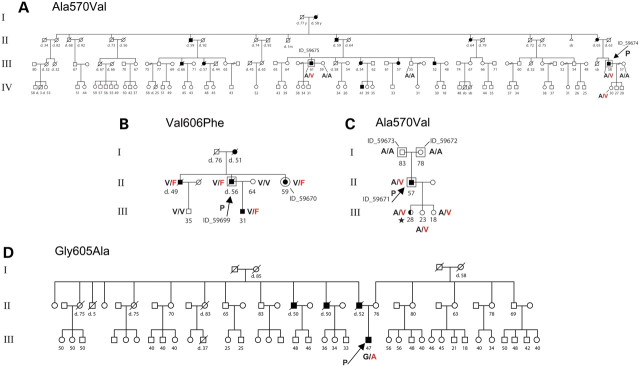
完成外显子组测序后,确定了一个额外的 ADCA-DN 家族,其中有四个已知的患者(图 1)。先证者DNMT1外显子 21 的 Sanger 测序在DNMT1的同一区域鉴定出第三个突变 p.Gly605Ala (RefSeq NM_001130823.1: c.1814C>G) 。有趣的是,所有三个突变都位于DNMT1蛋白内非常接近的空间位置(图 1)。使用 PolyPhen2 和 SIFT 评估了这些蛋白质变体的功能后果(所有突变都改变了在所有已检查的物种中的氨基酸),并且基因突变经过基因解码预测为具有破坏性。这些结果确定DNMT1为ADCA-DN 的致病基因。
ADCA-DN基因检测为什么可以避免后代及后胎再次患病?
{kind=link}
四个具有常染色体显性共济失调、耳聋和嗜睡症
(ADCA-DN) 的亲属中的DNMT1突变。(
ADCA-DN综合征的致病基因鉴定基因解码:靶向定位基因检测的科学依据
根据《人体基因序列变化及其疾病表征》,DNMT1是一种在人体内各种组织广泛表达的 DNA
甲基转移酶,在发育过程中维持甲基化模式,并通过直接结合HDAC2
佳学基因病案集中的三个突变位于蛋白质的外显子 21 中,突变改变在所有脊椎动物中高度保守的氨基酸(图
2)。
进一步证实该突变的致病性还来自最近发表的一个基因解码研究,研究称其他DNMT1突变(p.Asp490Glu、p.Pro491Tyr、p.Tyr495Cys)均位于DNMT1外显子
20的不同区域(图 2)。导致遗传性感觉丧失并伴有痴呆和听力丧失
(
DNMT1内的不同突变如何产生不同的迟发表型,并优先靶向选定的群体神经元,如hypocretin细胞或浦肯野细胞?一种可能是DNMT1通过
CpG
甲基化引起的基因沉默抵消了某些基因的过度表达,导致中枢和周围神经系统的某些神经元随着衰老而发生不需要的细胞聚集体的积累。这是患者脑脊液中为什么会出现tau
蛋白水平升高和痴呆症。关于共济失调,佳学基因解码的研究表明,减数分裂期间DNMT1活性的丧失会增加SCA1
在个别病例中,发作性睡病/hypocretin缺乏症是一种与 HLA-DQB1*06:02 (∼99%)
密切相关的散发性疾病。还发现了与 T 细胞受体 α (
最后,还值得注意的是,最近的一项全基因组关联研究在散发病例的P2RY11 (MIM 602697)
基因座中发现了发作性睡病相关多态性,该基因位于 (20-100
kb)
ADCA-DN综合征的致病基因之一的结构功能解析:基因解码基因检测的物质基础
https://www.jiaxuejiyin.com/uploads/allimg/2303/0040355156-1.jpg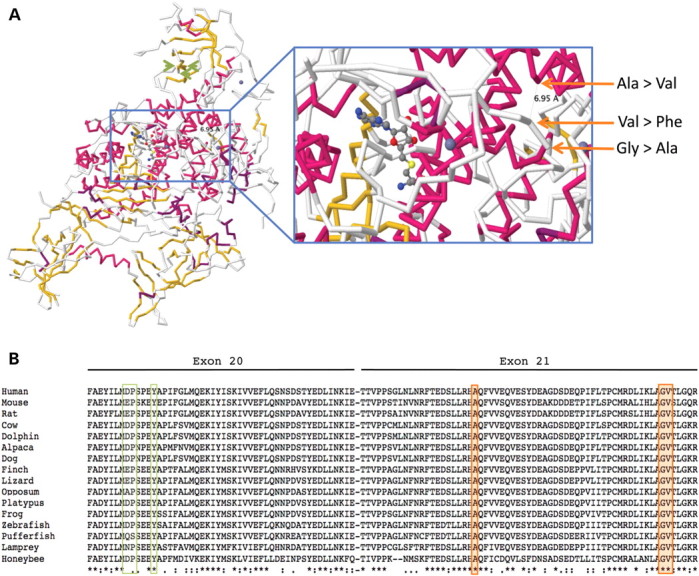{kind=link}
图 2:( A ) 小鼠DNMT1的蛋白质结构(PDB 登录号 3AV5,使用 Jmol_S 生成的图像)和 RFTS 域中新发现的氨基酸取代(橙色箭头)的位置。还显示了 Klein等人报告的氨基酸变化的位置。( 4 ),在蛋白质的 TS 结构域中(浅绿色箭头)。显示的底物是S -腺苷-l-同型半胱氨酸。( B ) 包含三个新突变(橙色框)和之前由 Klein等人报告的两个突变的区域的保护程度概述。( 4 ) (浅绿色方框)。
![]() 喜欢
喜欢
0
![]() 赠金笔
赠金笔