 加载中…
加载中…红外光谱学 维基百科,自由的百科全书
标签:
教育 |
分类: Research |
| 此条目需要补充更多来源。(2012年5月20日) 请协助添加来自可靠来源的引用以改善这篇条目。无法查证的内容会被提出异议而移除。 |
{kind=link}
{kind=link}
红外光谱学(英语:Infrared spectroscopy)是光谱学中研究电磁光谱红外部分的分支。它包括了许多技术,到目前为止最常用的是吸收光谱学。同所有的分光镜技术一样,它可以被用来鉴别一种化合物和研究样品的成分。红外光谱学相关表见于文献,方便查找。
目录 |
背景资料
红外光谱区是指波长在 0.8~1000 μm 范围内的电磁辐射区,习惯上再细分为近红外区(泛频区,0.8~2.5 μm,波数:14000–4000 cm−1)、中红外区(基本振动区,2.5~30 μm,4000–400 cm−1)和远红外区(转动区,30~1000 μm,400–10 cm−1)三个区域,其中中红外区是应用最多的区域。
理论
电磁光谱的红外部分根据其同可见光谱的关系,可分为近红外光、中红外光和远红外光。
远红外光(大约400-10
红外光谱法的工作原理是由于振动能级不同,化学键具有不同的频率。共振频率或者振动频率取决于分子等势面的形状、原子质量、和最终的相关振动耦合。为使分子的振动模式在红外活跃,必须存在永久双极子的改变。具体的,在波恩-奥本海默和谐振子近似中,例如,当对应于电子基态的分子哈密顿量能被分子几何结构的平衡态附近的谐振子近似时,分子电子能量基态的势面决定的固有振荡模,决定了共振频率。然而,共振频率经过一次近似后同键的强度和键两头的原子质量联系起来。这样,振动频率可以和特定的键型联系起来。
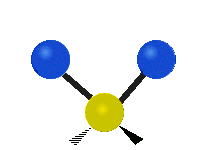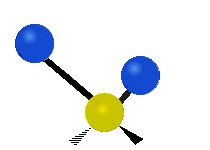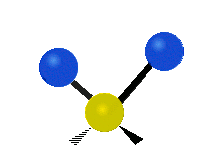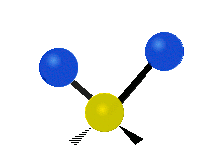
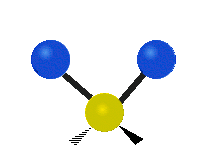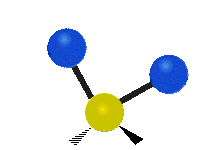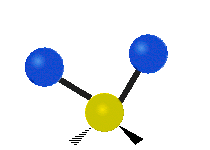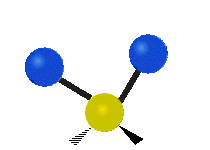
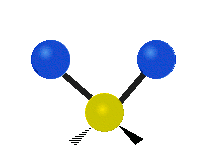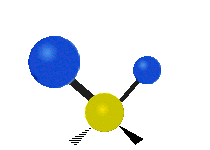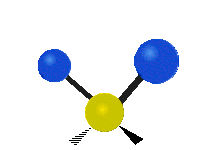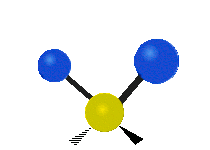
简单的双原子分子只有一种键,那就是伸缩。更复杂的分子可能会有许多键,并且振动可能会共轭出现,导致某种特征频率的红外吸收可以和化学组联系起来。常在有机化合物中发现的CH2组,可以以 “对称和非对称伸缩”、“剪刀式摆动”、“左右摇摆”、“上下摇摆”和“扭摆”六种方式振动(见下图)。
|
|
http://upload.wikimedia.org/wikipedia/commons/0/0c/Asymmetrical_stretching.gif维基百科,自由的百科全书" />
非对称伸缩
|
|
|
|
|
|
|
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
分子中的原子是以平衡点为中心在作小振幅的周期性振动,以双原子分子为例,它可以通过两端连接两个小球的弹簧谐振子来模拟,因此其简谐振动的频率可以通过下式得知:
- http://upload.wikimedia.org/wikipedia/zh/math/c/e/3/ce3b9105572f20cd898ded09758d00f6.png维基百科,自由的百科全书" /> ,其中 http://upload.wikimedia.org/wikipedia/zh/math/5/c/4/5c4a9ee0ca645652439e0246ae78c456.png维基百科,自由的百科全书" /> 为化学键的力常数,http://upload.wikimedia.org/wikipedia/zh/math/9/a/8/9a80f4c95fc7db6d0e0b7ae237795b71.png维基百科,自由的百科全书" /> 为折合质量。
{kind=link}
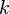{kind=link}
{kind=link}
这样还可求得分子的振动能 http://upload.wikimedia.org/wikipedia/zh/math/b/f/f/bffab2317681739dd86e4681b5b39622.png维基百科,自由的百科全书" /> :
{kind=link}
- http://upload.wikimedia.org/wikipedia/zh/math/5/0/6/5063a212117e518d00712789c2a6ba13.png维基百科,自由的百科全书" /> ,其中 http://upload.wikimedia.org/wikipedia/zh/math/a/7/9/a790ce61142c5e592046208969b9906f.png维基百科,自由的百科全书" /> ,称为振动量子数。
{kind=link}
{kind=link}
相邻振动能级间的能量差为 http://upload.wikimedia.org/wikipedia/zh/math/6/1/c/61cfb43d922748fa198b001d7e599410.png维基百科,自由的百科全书" /> 。谐振子的跃迁旋律为 http://upload.wikimedia.org/wikipedia/zh/math/d/3/2/d323af3dbf121cb75d9ca3105bd9ea57.png维基百科,自由的百科全书" /> ,所以双原子分子只能产生一条振动谱带。
{kind=link}
{kind=link}
但实际上双原子分子的振动位能曲线与谐振子还是有差别的,两者只是在 http://upload.wikimedia.org/wikipedia/zh/math/8/5/d/85d615cbb8327614ceea24190c49ae43.png维基百科,自由的百科全书" /> 较小的时候比较近似。真实分子的位能在原子间距离逐渐增大的时候,逐渐接近一恒定的解离能值。
{kind=link}
测量样品时,一束红外光穿过样品,个个波长上的能量吸收被记录下来。这可以由连续改变使用的单色波长来实现,也可以用傅立叶变换来一次测量所有的波长。这样的话,透射光谱或吸收光谱或被记录下来,显示出被样品红外吸收的波长,从而可以分析出样品中包含的化学键。
这种技术专门用在共价键的分析上,而且主要用于有机化学中。如果样品的红外活跃键少、纯度高,得到的光谱会相当清晰,效果好。更加复杂的分子结构会导致更多的键吸收,从而得到复杂的光谱。但是,这项技术还是用在了非常复杂的混合物的定性研究当中。
光谱图
红外吸收光谱图一般为 T-σ 曲线或 T-λ 曲线。纵坐标为透射比(T/%),吸收峰向下。横坐标为波数(σ/cm−1)或波长(λ/μm)。
吸收区域
-
主条目:红外光谱学相关表
{kind=link}
{kind=link}
4000~1350 cm−1 称为基频区,为化学键和官能团的特征振动频率区,可作为鉴定基团的依据。1350~650 cm−1 称为指纹区,与 C-C、C-O、C-X 单键的伸缩振动和分子骨架的弯曲振动有关,因各种单键强度大致相同,故这一区域的光谱非常复杂,适合于化合物的鉴别。
样品制备
- 气体——气体池
- 液体
- 液膜法——难挥发液体(bp>80°C)
- 溶液法——液体池
- 固体:
- 研糊法(液体石腊法)
- KBr压片法
- 薄膜法
典型方法
在有机分子中吸收键的总结
基团频率和特征吸收峰物质的红外光谱是其分子结构的反映,谱图中的吸收峰与分子中各基团的振动形式相对应。多原子分子的红外光谱与其结构的关系,一般是通过实验手段得到。这就是通过比较大量已知化合物的红外光谱,从中总结出各种基团的吸收规律。
C等,都有自己的特定的红外吸收区域,分子的其它部分对其吸收位置影响较小。通常把这种能代表及存在、并有较高强度的吸收谱带称为基团频率,其所在的位置一般又称为特征吸收峰。º实验表明,组成分子的各种基团,如O-H、N-H、C-H、C=C、C=OH和C
一、基团频率区和指纹区
(一)基团频率区 中红外光谱区可分成4000 cm-1 ~1300 cm-1和1800cm-1 (1300 cm-1 )~ 600 cm-1两个区域。最有分析价值的基团频率在4000 cm-1 ~ 1300 cm-1 之间,这一区域称为基团频率区、官能团区或特征区。区内的峰是由伸缩振动产生的吸收带,比较稀疏,容易辨认,常用于鉴定官能团。 在1800 cm-1 (1300 cm-1 )~600 cm-1 区域内,除单键的伸缩振动外,还有因变形振动产生的谱带。这种振动与整个分子的结构有关。当分子结构稍有不同时,该区的吸收就有细微的差异,并显示出分子 特征。这种情况就像人的指纹一样,因此称为指纹区。指纹区对于指认结构类似的化合物很有帮助,而且可以作为化合物存在某种基团的旁证。基团频率区可分为三 个区域:LT7U N基吸收比较强而尖锐。若分子中含有O原子,且O原子离-Cº键或芳香核共轭时,该峰位移到2220~2230 cm-1附近。若分子中含有C、H、N原子, -C N基的吸收越弱,甚至观察不到。N基越近, -C º 1900~1200 cm-1为双键伸缩振动区 该区域重要包括三种伸缩振动:
- C=O伸缩振动出现在1900~1650 cm-1 ,是红外光谱中很特征的且往往是最强的吸收,以此很容易判断酮类、 醛类、酸类、酯类以及酸酐等有机化合物。酸酐的羰基吸收带由于振动耦合而呈现双峰。
- C=C伸缩振动。烯烃 的C=C伸缩振动出现在1680~1620 cm-1 ,一般很弱。单核芳烃的C=C伸缩振动出现在1600 cm-1和1500 cm-1附近,有两个峰,这是芳环的骨架结构,用于确认有无芳核的存在。
- 苯的衍生物的泛频谱带,出现在2000~1650 cm-1范围, 是C-H面外和C=C面内变形振动的泛频吸收,虽然强 度很弱,但它们的吸收面貌在表征芳核取代类型上是有用的。
(二)指纹区d 1. 1800(1300)~900 1375»cm-1区域是C-O、C-N、C-F、C-P、C-S、 P-O、Si-O等单键的伸缩振动和C=S、S=O、P=O等双键的伸缩振动吸收。 其中 C-H对称弯曲振动,对识别甲基十分有用,C-O的伸缩振动在1300~1000 cm-1dcm-1的谱带为甲基的 ,是该区域最强的峰,也较易识别。 900~650 cm-1区域的某些吸收峰可用来确认化合物的顺反构型。 例如,烯烃的=C-H面外变形振动出现的位置,很大程度上决定于双键的取代情况。对于RCH=CH2结构,在990 cm-1和910 cm-1出现两个强峰;为RC=CRH结构是,其顺、反构型分别在690 cm-1和970 cm-1出现吸收峰,可以共同配合确定苯环的取代类型。
二、常见官能团的特征吸收频率
三、影响基团频率的因素
基团频率主要是由基团中原子的质量和原子间的化学键力常数决定。然而,分子内部结构和外部环境的改变对它都有影响,因而同样的基团在不同的分子和不 同的外界环境中,基团频率可能会有一个较大的范围。因此了解影响基团频率的因素,对解析红外光谱和推断分子%( 结构都十分有用。 影响基团频率位移的因素大致可分为内部因素和外部因素。 内部因素:
- 电子效应 包括诱导效应、共轭效应和中介效应,它们都是由于化学键的电子分布不均匀引起的。
- 诱导效应(I 效应) 由于取代基具有不同的电负性,通过静电诱导作用,引起分子中电子分布的变化。从而改变了键力常数,使基团的特征频率发生了位移。 例如,一般电负性大的基团或原子吸电子能力强,与烷基酮羰基上的碳原子数相连时,由于诱导效应就会发生电子云由氧原子转向双键的中间,增加了C=O键的力 常数,使C=O的振动频率升高,吸收峰向高波数移动。随着取代原子电负性的增大或取代数目的增加,诱导效应越强,吸收峰向高波数移动的程度越显著。
- 中介效应(M效应)当含有孤对电子的原子(O、S、N等)与具有多重键的原子相连时,也可起类似的共轭作用,称为中介效应。由于含有孤对电子的原 子的共轭作用,使C=O上的电子云更移向氧原子,C=O双键的电子云密度平均化,造成C=O键的力常数下降,使吸收频率向低波数位移。 对同一基团,若诱导效应和中介效应同时存在,则振动频率最后位移的方向和程度,取决于这两种效应的结果。当诱导效应大于中介效应时,振动频率向高波数移 动,反之,振动频率向低波数移动。
- 氢键的影响氢键的形成使电子云密度平均化,从而使伸缩振动频率降低。游离羧酸的C=O键频率出现在1760 cm-1 左右,在固体或液体中,由于羧酸形成二聚体, C=O键频率出现在1700 cm-1 。 分子内氢键不受浓度影响,分子间氢键受浓度影响较大。
- 振动耦合 当两个振动频率相同或相近的基团相邻具有一公共原子时,由于一个键的振动通过公共原子使另一个键的长度发生改变,产生一个“微扰”,从而形成了强烈的振 动! 相互作用。其结果是使振动频率发生感变化,一个向高频移动,另一个向低频移动,谱带分裂。振动耦合常出现在一些二羰基化合物中,如,羧酸酐。
- Fermi共振 当一振动的倍频与另一振动的基频接近时,由于发生相互作用而产生很强的吸收峰或发生裂分,这种现象称为Fermi共振。外部因素 外部因素主要指测定时物质的状态以及溶剂效应等因素。 同一物质的不同状态,由于分子间相互作用力不同,所得到光谱往往不同。 分子在气态时,其相互作用力很弱,此时可以观察到伴随振动光谱的转动精细结构。 C-H为1742n液态和固态分子间作用力较强,在有极性基团存在时,可能发生分子间的缔合或形成氢键,导致特征吸收带频率、强度和形状有较大的改变。例 如,丙酮在气态时的 cm-1 ,而在液态时为1718 cm-1 。 在溶液中测定光谱时,由于溶剂的种类、溶剂的浓度和测定时的温度不同,同一种物质所测得的光谱也不同。通常在极性溶剂中,溶质分子的极性基团的伸缩振动频率随溶剂极性的增加而向低波数方向移动,并且强度增大。因此,在红外光谱测定中,应尽量采用非极性的溶剂。
运用
傅立叶变化红外光谱学
目前几乎所有的红外光谱仪都是傅立叶变换型的。色散型仪器的主要不足是扫描速度慢,灵敏度低,分辨率低。因此色散型仪器自身局限性很大。
傅立叶变换红外光谱仪的构成
(图5.7)中,光源发出的光被分束器分为两束,一束经反射到达动镜,另一束经透射到达定镜。两束光分别经定镜和动镜反射再回到分束器。动镜以一恒 定速度vm作直线运动,因而经分束器分束后的两束光形成光程差d,产生干涉。干涉光在分束器会合后通过样品池,然后被检测。傅立叶变换红外光谱仪的检测器 有TGS,MCT等。
傅立叶变换红外光谱的基本原理
傅立叶变换红外光谱仪的核心部分是迈克耳孙干涉仪, 其示意图如(图5.8)所示。动镜通过移动产生光程差,由于vm一定,光程差与时间有关。光程差产生干涉信号,得到干涉图。光程差d = 2d,d代表动镜移动离开原点的距离与定镜与原点的距离之差。由于是一来一回,应乘以2。若d=0,即动镜离开原点的距离与定镜与原点的距离相同,则无相 位差,是相长干涉;若d=l/4,d=l/2 时,位相差为l/2,正好相反,是相消干涉;d=l/2,d=l 时,又为相长干涉。总之,动镜移动距离是l/4的奇数倍,则为相消干涉,是l/4的偶数倍,则是相长干涉。因此动镜移动产生可以预测的周期性信号。
参考资料
- 华中师范大学、陕西师范大学、东北师范大学 编.《分析化学》下册 第三版.北京:高等教育出版社,2001年.ISBN 978-7-04-009291-2.
![]() 喜欢
喜欢
0
![]() 赠金笔
赠金笔