 加载中…
加载中…PICRUSt功能预测 瞬间提高微生物多样性研究性价比
标签:
微生物高通量测序picrust功能预测16s |
分类: sci攒人品ing |
https://wenku.baidu.com/view/294dae53b207e87101f69e31
众所周知,同是微生物DNA水平的测序策略,宏基因组测序可以更好地分析群落功能,16S等多样性测序则更多只能在物种组成和多样性层面去解答微生物群落结构问题。如果要开展宏基因组测序,价格是16S的十几倍,对于经费不充足的实验室来说是一个巨大的考验。那我们到底有没有机会利用较便宜的16S测序来开展功能研究,使文章内容更为丰富,研究更具性价比呢?
答案是有的,今天我们给大家介绍一款名为PICRUSt的软件。这款软件第一次见刊于2013年的Nature Biotechnology文章当中,短短3年时间就有600多个引用量,相当不错。
http://img.mp.itc.cn/upload/20161111/090ca59a8e6c47959f07f4a43f2470ae.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
下面详细介绍一下这款软件~
预测原理
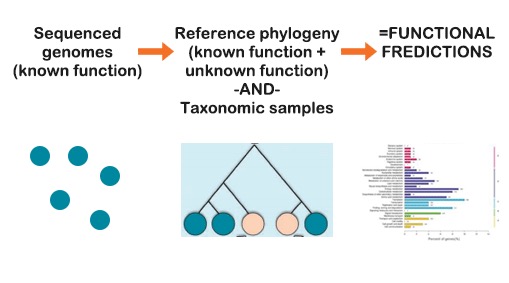
软件原理很简单,它通过搭建一个“物种—基因”的关系网,从而通过OTU预测群落的基因类型与数量,最后达到功能分析的目的。具体可分为三个阶段:
1
构建“物种—基因”关系网
网络构建需要两份重要的文件作为支持:
-
已有参考基因组细菌或古细菌中每个基因家族的基因数量(通过IMG数据库获得);
-
和通过物种所构建的进化树(通过greengene数据库获得)。
通过这两个数据,软件就可以建立一个已知物种基因信息的进化树。
2
实现物种输入到基因输出的转化
前面已经根据已知的物种建立起一个带有基因信息(种类和数量)的进化树,通过未知物种的序列信息寻找其在进化树中的亲缘物种,从而根据亲缘物种的基因信息预测未知物种的基因信息。由于微生物的变异速度极快,大量的HGT和基因丢失现象可能会使这种预测方式会存在一定的偏差,但由于近源物种间的主要基因信息还是非常接近,因此预测结果可靠性还是极高(下文会有证明)。
3
进行基因功能注释
PICRUSt完成宏基因组的预测之后,可以利用软件结合KEGG,COG和Pfam三大数据库进行注释,从而赋予基因信息生物学意义。
http://img.mp.itc.cn/upload/20161111/3969033c0db34afab513c66f6b43a313_th.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
如何操作
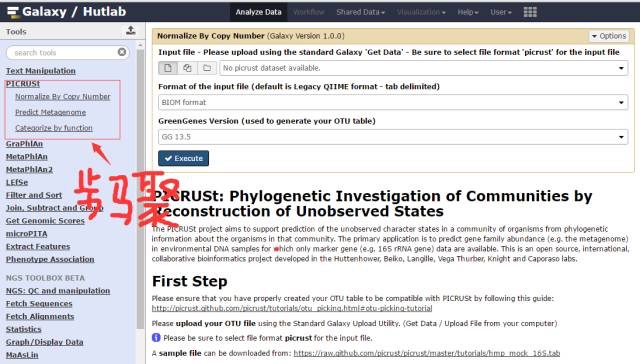
打开网址:http://picrust.github.io/picrust/,可以下载到本地安装,也可以在线操作。打开软件,输入OTU数据表格,选择适当的参数,例如选择tab格式还是BIOM格式的输入文件,选择进化树调用数据库是Greengene的哪个版本,选择注释数据库是KEGG还是COG等常用参数,点击执行后即可实现基因预测。具体操作细节可查看网络帮助文件。
http://img.mp.itc.cn/upload/20161111/a94a874a931c49ba99f391f0edd3ea19_th.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
预测结果可靠性评估
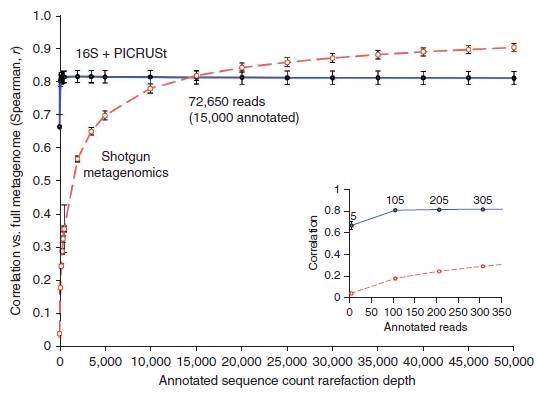
从下图可以发现,利用16S + PICRUSt的组合(蓝线)与宏基因组测序(红线)所得到的宏基因组数据进行比较发现,16S + PICRUSt组合所得到的宏基因组数据能够极高地接近真实水平(r > 0.8)当测序深度较低的时候,16S + PICRUSt组合的效果甚至比宏基因组测序的更好。因此,PICRUSt软件能在有限的16S测序数据中较高地还原样本的功能信息。
http://img.mp.itc.cn/upload/20161111/6a40320e1d61427d895ad48141c3eb13_th.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
(数据来源于Langille et. al.)
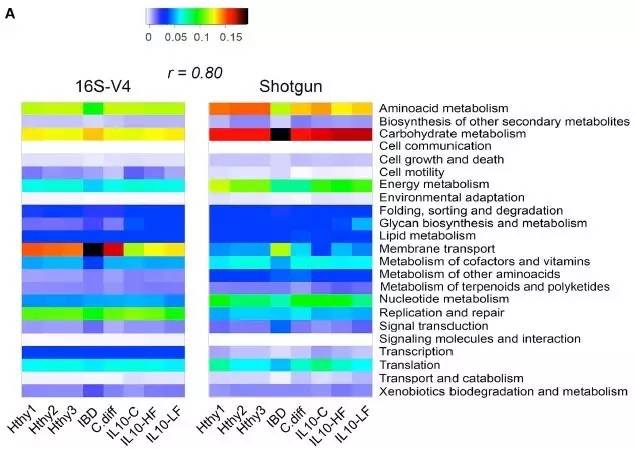
从下图中16S的PICRUSt基因通路预测(左图)和宏基因组测序的通路预测(右图)的对比,发现两者的相似度极高(r=0.8)。同时,针对不同样本,PICRUSt预测都展示出与宏基因组基本相似的结果,因此相比于价格极高的宏基因组测序,16S + PICRUSt组合在群落功能研究上展现出极高的性价比。
http://img.mp.itc.cn/upload/20161111/2ae9c340b9764d77938cfc645894ab7b_th.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
(数据来源于Jovel et. al.)
小结
PICRUSt软件能够很好地搭建从物种到基因功能的桥梁,通过生物信息算法很好地挖掘微生物群落研究能信息,在有限的资源下是16S研究的一个很好的应用手段。基迪奥的16S多样性也提供这种分析,追求极致性价比的老师可以随时联系我们。
参考文献:
【1】Langille, Morgan GI,
et al. "Predictive functional profiling of microbial communities
using 16S rRNA marker gene sequences."
【2】Jovel,
Juan, et al. "Characterization of the Gut Microbiome Using 16S or
Shotgun me tagenomics."
{kind=link}
PICRUSt全称为
Phylogenetic Investigation of Communities
by Reconstruction of Unobserved States
好了
全称不重要
发音很重要
:pie crust
装X从发音正确开始
分析上您有两种选择:
一、小文艺的选择是,软件本地化操作(但需要linux基础);
二、直接在线分析,简单粗暴。
PICRUSt的原理基于已测细菌基因组的16S rRNA全长序列,推断它们的共同祖先的基因功能谱,对Greengenes数据库中其它未测物种的基因功能谱进行推断,构建古菌和细菌域全谱系的基因功能预测谱,最后,将测序得到的菌群组成“映射”到数据库中,对菌群代谢功能进行预测。
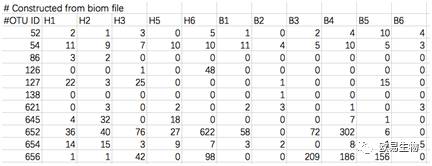
您需要的是一份BIOM格式的OTU文件或者转换为txt二维矩阵的OTU丰度表格。前者格式比较小众,且相对复杂不做介绍,后者长这样:
http://img.mp.itc.cn/upload/20170103/2763ef046dd04516abc430deef170920.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
只要您在欧易做的微生物多样性项目,我们报告都会提供这两个文件的啦。有问题欢迎随时咨询。
step1
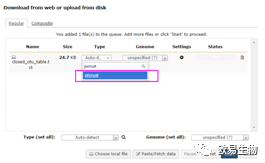
打开网址
Type记得一定要点选 “picrust”, 然后点击下方的“Start”。
http://img.mp.itc.cn/upload/20170103/c393d596f304486585e0b9189a2062d9.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
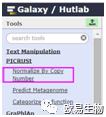
step2
点击
Normalize By Copy Number, 右边部分转着转着就绿了,绿了就OK了。
http://img.mp.itc.cn/upload/20170103/1899b9d0451f49e290749d610842ec5c.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
http://img.mp.itc.cn/upload/20170103/5c03f4956ca24f1099401f5f4ad631ab.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
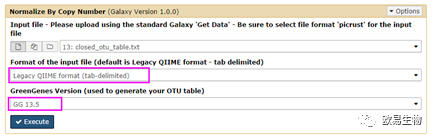
选择“Legacy QIIME format(tab)”以及Greengenes数据库版本“GG 13.5“。
step3
点击左上方“Predict Metagenome”,这一步会根据step2得到预测出来的KEGG信息,储存在BIOM格式文件,下一步就是对这种小众格式文件的大众可视化了。在预测的分类中,按需选择“KEGG Orthologs”、“COG”或者“Rfam”。
http://img.mp.itc.cn/upload/20170103/ea3b8c94a06f40e09763ba1bd9616ca5.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
step4
点击左上方的“Categorize by function”,就得到对应的结果啦。
得到了KEGG三个层级的信息。
http://img.mp.itc.cn/upload/20170103/2a0827519eac4e889a005f05dee13b19.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
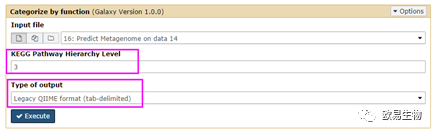
“KEGG Pathway Hierarchy Level“有三种层级可选;而“Type of output”建议就选择人类能看得懂的大众格式”Legacy QIIME format(tab)“。转绿后,点击保存可将结果下载。可自行重命名为“picrust.txt”。
http://img.mp.itc.cn/upload/20170103/5700f5c2056b4d49988d3ec90874f675.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
结果长这样:
http://img.mp.itc.cn/upload/20170103/8f97a8465d0f4a3797347f3c3918bb5f.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
第一列为KEGG第三个层级分类,最后一列为分号分隔的L1-L3三个层级详细分类。数字为样本中可能与该功能相关的reads拷贝数。
得到这表格信息后,数据能怎么用呢?多了去啦,比如组间t-test检验、heatmap聚类、PCA......。
基于KEGG的T-test检验:
http://img.mp.itc.cn/upload/20170103/983360fa16b84b2eb9d18097640f52e1.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
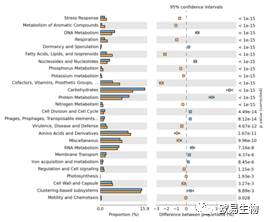
上图为比较不同分组样本的 KEGG pathway, 并筛选出具有显著性组间差异的 pathway,其中蓝色代表一个分组,橘黄色代表另外一个分组。左边横柱状图代表富集在该 KEGG 的 reads 数目,右边为 corrected p 值。
说到KEGG的t-test检验,笔者不得不再甩出一个包袱,下次再给大家分享一个超级好用,windows就可操作的软件:
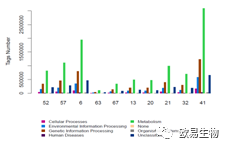
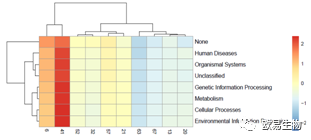
基于KEGG的heatmap分析以及Barplot分析:
http://img.mp.itc.cn/upload/20170103/7a82266dc22c489b998ac707a28db95f.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
http://img.mp.itc.cn/upload/20170103/657fa8489fe94e70a4f86fef4cadaf73.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
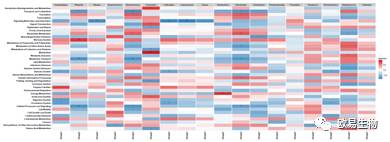
以及差异物种和KEGG预测通路相关性分析:
http://img.mp.itc.cn/upload/20170103/435c4d0a58544c1f8ae33db08cdebb81.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
上图中根据pearson相关性计算差异的种属和预测出KEGG通路的相关性。深红代表较高正相关,深蓝代表较高负相关,***代表相关性p value<0.001, **代表相关性p value在0.001~0.01之间, *代表相关性p value在0.01和0.05之间。
好啦,干货讲完,
来点更实在的重磅消息:
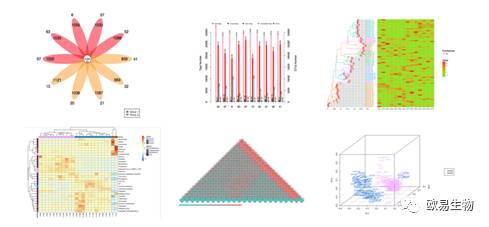
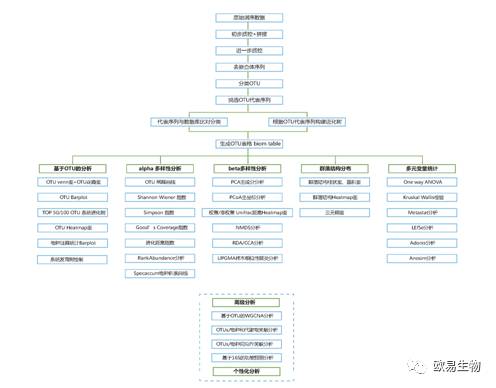
欧易生物微生物多样性分析全线升级啦!!!!
不仅分析上加质加量不加价,图形可视化也让文艺小清新们欲罢不能。
http://img.mp.itc.cn/upload/20170103/7e1e705f45c043a9b5993f16450db819.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
http://img.mp.itc.cn/upload/20170103/bc915f58f61446ffbbbaabdc41de7c83_th.jpg瞬间提高微生物多样性研究性价比" />
{kind=link}
看到这里有没有心动呢?如果有任何疑惑或者需求,欢迎各位老师邮件或者致电详询哦。
1Langille MG, Zaneveld J, Caporaso JG,
et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 2013, 31(9): 814-821.
![]() 喜欢
喜欢
0
![]() 赠金笔
赠金笔